New article published in JCTC: « Lambda-ABF: Simplified, Accurate and Cost-effective Alchemical Free Energy Computations« by
L. Lagardère, L. Maurin, O. Adjoua, K. El Hage, P. Monmarché, J.-P. Piquemal, J. Hénin, J. Chem. Theory. Comput., 2024, 20 (11), 4481-4498
https://doi.org/10.1021/acs.jctc.3c01249
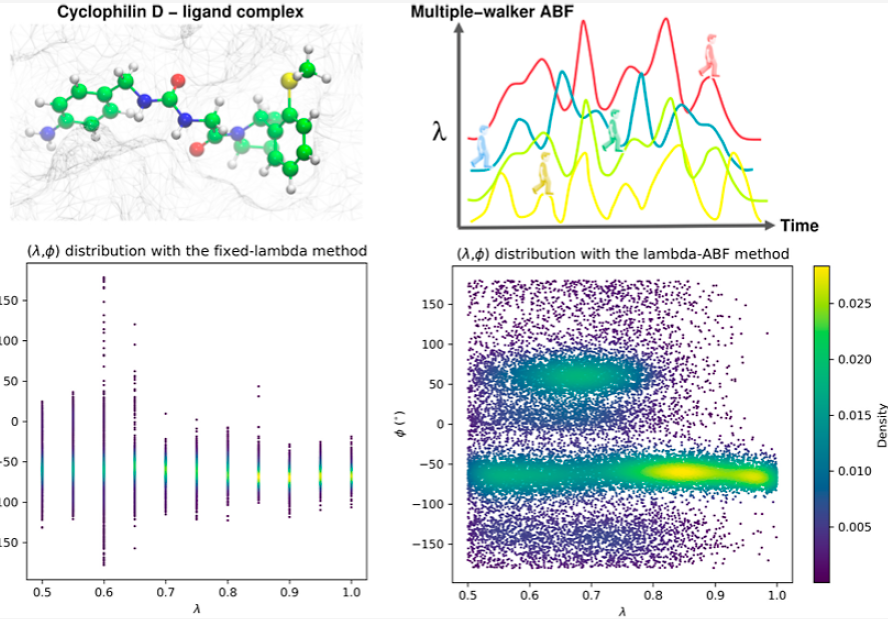
We introduce the lambda-Adaptive Biasing Force (lambda-ABF) method for the computation of alchemical free-energy differences. We propose a software implementation and showcase it on biomolecular systems. The method arises from coupling multiple-walker adaptive biasing force with λ-dynamics. The sampling of the alchemical variable is continuous and converges toward a uniform distribution, making manual optimization of the λ schedule unnecessary. Contrary to most other approaches, alchemical free-energy estimates are obtained immediately without any postprocessing. Free diffusion of λ improves orthogonal relaxation compared to fixed-λ thermodynamic integration or free-energy perturbation. Furthermore, multiple walkers provide generic orthogonal space coverage with minimal user input and negligible computational overhead. We show that our high-performance implementations coupling the Colvars library with NAMD and Tinker-HP can address real-world cases including ligand–receptor binding with both fixed-charge and polarizable models, with a demonstrably richer sampling than fixed-λ methods. The implementation is fully open-source, publicly available, and readily usable by practitioners of current alchemical methods. Thanks to the portable Colvars library, lambda-ABF presents a unified user interface regardless of the back-end (NAMD, Tinker-HP, or any software to be interfaced in the future), sparing users the effort of learning multiple interfaces. Finally, the Colvars Dashboard extension of the visual molecular dynamics (VMD) software provides an interactive monitoring and diagnostic tool for lambda-ABF simulations.